Sickle Cell Disease

Take home messages
- Sickle cell disease is an autosomal co-dominant congenital haemoglobinopathy
- The two main problems are vaso-occlusive crises and haemolysis
- Avoid hypoxia, pain, dehydration, hypothermia and acidosis if you're anaesthetising someone with sickle cell disease
Podcast episode
Crisis time
If you've been called to see a patient in acute crisis
Clearly the gold standard answer is 'refer to the acute pain team', but often as the weekend CEPOD anaesthetist - you are the pain team.
Here's our quick reference list of things to think about
- Is it something else? - appendicitis, cholecystitis, stroke, pneumonia?
- Pain control - paracetamol, ibuprofen, oramorph, nasal fentanyl, PCA
- Oxygen - give more than usual for at least 24 hours
- Fluids - resuscitation and maintenance
- Keep them warm
- Avoid acidosis
- Who else needs to know? - haematology/paediatrics
- Check their sickle cell passport if they have one
- Laxatives! - kids become spectacularly constipated on opioids
Pathophysiology
As you'll remember immediately from medical school, haemoglobin has four polypeptide chains, each covalently bound to a haem moiety.
Within this haem moiety we find a porphyrin ring at the centre of which resides an iron atom in its ferrous state (Fe 2+)
Some types of haemoglobin to know
- HbA1 - 96% of adult haemoglobin - α2β2
- HbA2 - 2-3% of adult haemoglobin - α2δ2
- HbF - Foetal - α2γ2
- HbS - Sickle cell
- HbH - severe alpha thalassaemia - β4
As you might imagine - there are many more.
- α subunits are encoded by chromosome 16
- β or non-α subunits are encoded by chromosome 11
What happens in sickle cell
Sickle cell is a congenital haemoglobinopathy as a result of a mutation on chromosome 11.
This produces HbS.
- Homozygotes are HbSS and have sickle cell disease
- Heterozygotes are HbAS and have sickle cell trait
The problems arise because HbS is prone to polymerising at low partial pressures of oxygen, forming structures called tactoids, which are far less cool than they sound.
This causes the red blood cell to lose its flexibility, making them stiff and fragile - hence they're said to 'sickle'.
HbAS can also sickle, but only at very low partial pressures of oxygen, which don't tend to occur in usual clinical practice, so sickle cell trait patients don't generally suffer from the same symptoms as HbSS.
The effects of sickling
- Increased blood viscosity
- Microthrombi in the peripheral circulation
- Vasoocclusive crises
- Haemolytic anaemia
- Progressive inflammatory vascular endothelial damage
Sickling red cells last around 5-15 days, rather than the usual 120.
What factors can induce sickling?
- Hypoxia - HbSS will sickle at 85%, HbAS will sickle at 40%
- Hypothermia
- Acidosis
- Dehydration
- Pooling of blood or stasis
- Low cardiac output
- Hypovolaemia
Effects on the body
You might get asked about the systemic effects of sickle cell disease on the body, and as always it is best to break it down by organ system:
Respiratory
Symptoms of recurrent pulmonary infarction:
- Dyspnoea
- Pleuritic chest pain
- Cough
- Haemoptysis
Cardiovascular
- Cardiomegaly - due to anaemia
- Pulmonary hypertension
- Congestive heart failure
Neurological
- TIA
- CVA
- Haemorrhage
Haematological
- Anaemia
- Haemolysis
- Parvovirus B19-induced hypoplastic crisis
- Aplastic anaemia
Musculoskeletal
- Frontal bossing and maxillary hypertrophy in extreme cases
- Skin ulcers
GI and GU
- Renal failure
- Priapism
- Splenomegaly
- Splenic infarction
- Gallstones
- Hepatomegaly
Airway
- Sleep apnoea due to tonsillar and adenoid hypertrophy
Eye
- Microvascular retinopathy
- Retinal detachment
- Vitreous haemorrhage
Pain crises
Apart from a patient with sickle cell disease (or trait) passing through your anaesthetic room for an unrelated operation, the other time you'll tend to find yourself in front of a patient with sickle cell as an anaesthetist is during an acute pain crisis.
These cruel repeated episodes of severe unrelenting pain can be triggered by a number of stresses including dehydration, pain, stress and even changes in altitude, and the ward team may well call upon the anaesthetists for help with specialist methods of pain control.
The priority of course is analgesia, but it is also crucial to ensure you're not missing something else that may be bubbling away in the background.
Is the pain normal for them during a crisis?
Sickle cell patients know their pain very well indeed, and can often predict when a crisis is going to take hold with remarkable accuracy.
The following scenarios should make you feel very differently:
- "I always get this pain in my back and lower abdomen, it lasts for a few hours and I usually need IV morphine to get on top of it"
- "She's complaining of abdominal pain but usually her pain is higher up in the back - this time seems different"
Sickle cell crisis is a great mimic, but it can also conceal other underlying diagnoses, which you need to be on the lookout for.
Remember an underlying pathology can also trigger a pain crisis. Just to make life easier for everyone involved.
Things to think about
- Bone infarction
- Osteomyelitis
- Appendicitis
- Cholecystitis
- Constipation due to opioids
- Pulmonary crisis
- Pneumonia
- Meningitis
- Cerebral infarction
Things to monitor
- Temperature
- Focal signs of infection
- Heart rate
- Blood pressure
- Respiratory rate
- Rigors
- White cell count
- Cap refil time
If you're in any doubt, escalate your concerns and encourage investigation of possible underlying pathologies - we don't want to leave a 15 year old in crisis with a ruptured appendix now do we.
Pain control options
- Paracetamol
- Ibuprofen
- Oramorph
- Nasal fentanyl
- PCA or IV morphine
- MST once a baseline requirement is calculated
With regards to dosing, your trust will most likely have a pro-forma with the approved doses by weight or age for sickle cell patients, like this incredibly helpful one from Norfolk and Norwich University Hospitals that we found online.
Non-pharmacological
- Keep them warm
- Reassurance
- Parental presence
- Distraction
Paw patrol isn't going to reduce the pain of an acute sickle crisis but it's markedly more tolerable than staring at a blank wall.
Testing for it
The sickledex test is an easily performed bedside test which reveals the presence of HbS, but does not differentiate between the heterozygous (sickle cell trait) and the homozygous state.
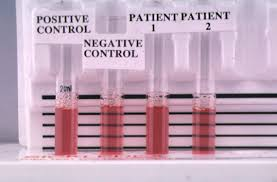
How can you test for it?
- Sickledex test
- Exposes red cells to sodium metabisulphite
- This will cause sickling in HbS but won't distinguish between HbSS and HbAS
- High performance liquid chromatography and electrophoresis are other options
- If Sickledex is positive then HPLC or electrophoresis required to confirm disease vs trait
How it affects your anaesthetic
What you'll notice in this section is that most of the management is exactly what you'd do for any other patient, but you just need to do it even better.
In the bad old days, perioperative mortality in unwell sickle cell patients hovered around 30-40%, but it's mercifully more like 1% now that we've figured out what we're supposed to do.
Oxygen
The most important feature is to avoid hypoxia. While this is important in all patients, it is especially crucial in order to avoid sickling in HbSS carriers.
Hypoxia is exceedingly poorly tolerated in these patients.
For those alveolar gas equation aficionados among you - slight hyperventilation can also increase arterial oxygen tensions if needed.
Hydration
Again - all patients need to be adequately hydrated - but hypovolaemia is bad news for those with shapeshifting erythrocytes.
Transfusion
Crossmatching is going to take longer in these patients, so make sure group and saves and crossmatching requests are done well in advance wherever possible.
You can also consider prophylactic transfusion to achieve two things:
- aim for an Hb >100
- try and replace as much of their sickly red cells with non-sickly ones
Clearly this carries all the requisite risks associated with elective blood transfusion and should probably be discussed on an individualised basis with haematology.
Other things to think about
- Avoid hypothermia - increased risk of tissue hypoxia and sickling
- Avoid infection - prophylactic antibiotics
- If they've had previous transfusions (especially in other countries) think about the risk of blood-borne virus transmission
Patients with sickle cell disease tend to have functional hyposplenism, so they're usually on some form of long-term antibiotic.
Fluid management
It is likely that a patient - particularly a child - in severe pain hasn't been drinking enough. Furthermore sickle cell patients often have an element of reduced nephron function already. This results in dehydration, increased blood viscosity and more sickling.
Oral fluids are always preferable, but if needed to avoid dehydration then IV replacement can be started. Choice of fluid varies between hospitals, but as long as it's vaguely balanced with some sugar and you're monitoring potassium then it doesn't matter too much.
For Adults:
- Encourage oral fluids where possible
- Aim for a total fluid intake of 70-90 ml/kg body weight over 24 hours as maintenance
- Hypovolaemia or clinical dehydration should be treated in addition to this
- Start a fluid balance chart
- It is very important to avoid fluid overload, as hypoxia from pulmonary oedema can be catastrophically bad
- Avoid central lines unless absolutely needed
It will not surprise you that viscous, sickly blood rather enjoys setting up camp on indwelling lines.
For a child for 24 hours:
- 100ml/kg for the first 10kg
- 50ml/kg for the next 10kg
- 20ml/kg thereafter
So a 19kg child would require 10x100 + 9x50 = 1450 ml over 24 hours.
Life threatening complications
The majority of the time, pain crises will require pain control and rehydration but not a whole lot more.
But you need to be on the lookout for the really bad stuff so you can spot and manage it in good time.
Stroke
- Can present with headache, weakness or convulsions
- Think about meningitis
- Anticonvulsants as required
- CT and LP
- May need urgent exchange transfusion
Severe anaemia
- Due to haemolysis or splenic/hepatic sequestration
- Suspect if sudden drop in Hb of more than 20 g/litre
- Can present with cardiovascular collapse as well
- Often due to parvovirus B19 infection
- Needs urgent transfusion and antibiotics
Girdle syndrome and abdominal pain
- Can be due to mesenteric occlusion
- Need to rule out other causes of acute abdominal pain
- Cholecystitis more common in context of haemolysis
- Girdle syndrome (mesenteric sickling) can present with distension and vomiting
Useful Tweets and Resources
25-year-old, sickle cell disease, evolving CXRs. Day 1 looks clear, but what's next? Can you track the changes? #MedTwitter #MedEd #FOAMed https://t.co/nhCYooXctc pic.twitter.com/6OlxtX0hVC
— LITFL (@LITFLblog) January 5, 2024
References and Further Reading



Primary FRCA Toolkit
While this subject is largely the remit of the Final FRCA examination, up to 20% of the exam can cover Primary material, so don't get caught out!
Members receive 60% discount off the FRCA Primary Toolkit. If you have previously purchased a toolkit at full price, please email anaestheasier@gmail.com for a retrospective discount.

Discount is applied as 6 months free membership - please don't hesitate to email Anaestheasier@gmail.com if you have any questions!
Just a quick reminder that all information posted on Anaestheasier.com is for educational purposes only, and it does not constitute medical or clinical advice.