Clearance and Elimination
Take home messages
- Clearance is the volume of plasma completely and irreversibly cleared of drug per minute
- The half life is the time taken for the plasma concentration to drop to half of any initial value
- Redistribution is not elimination
Podcast episode
Pharmacokinetics
It's always a magical feeling when you see the milk of amnesia disappear into the cannula; the patient's eyes fluttering closed just thirty seconds later.
Clearly the drug has made its way from arm - or foot or wherever you've finally managed to cannulate - to brain where it then exerted its mysterious pharmacodynamic effects.
But what else is going on?
Where else does it go? How much actually reaches the brain? What happens afterwards? How much is too much and when should I give more? - these are questions of pharmacokinetics, or what the body does with the drug once it's inside.
There are four aspects to pharmacokinetics that you should always mention in any viva question on the topic:
- Absorption
- Distribution
- Metabolism
- Elimination
In this post we're looking at the last bit - the elimination that so often gets examined in the Primary FRCA exam.
Buckle up.
The equations you need to be able to recite
- Rate of elimination (mg/min) = Concentration (mg/ml) x clearance (ml/min)
- Volume of distribution (ml) = Dose (mg) / Concentration (mg/ml)
- Elimination rate constant (k) = 1/ time constant (min)
- Half life (T1/2) = 0.693 x time constant (min)
- Clearance (ml/min) = Volume of distribution (ml) x k (/min)
- Clearance = dose / Area under curve (concentration/time)
- Loading dose = Volume of distribution x target concentration
- Maintenance infusion rate = target concentration x clearance
What is elimination?
Let's start with a very simple definition.
Elimination is the irreversible removal of drug from the plasma, through one or both of the following mechanisms:
- Physical removal from the body (excretion)
- Metabolism into inactive breakdown products
Just note that redistribution to other bits of the body does not count as elimination, because although the molecule has left the plasma, it is very much coming back to haunt us later (it's not irreversible), as we'll explain in a bit.
Most polar or water-soluble drugs are excreted by the kidneys, and some larger molecules can be excreted into the bile.
The kidneys aren't very good at dealing with fat-soluble, non-polar drugs, so these need to be metabolised into a polar, soluble molecule first, usually by the liver, where they can undergo phase 1 and phase 2 reactions.
Phase 1 reactions
- Oxidation
- Reduction
- Hydrolysis
These reactions generally result in addition or activation of a functional group, such as OH, COOH or NH2.
They also generally make the molecule more water soluble.
Phase 2 reactions
- Glucuronidation
- Sulphation
- Acetylation
- Methylation
- Conjugation
These reactions generally make the molecule much more water soluble.
Things to remember about Phase 1 and 2 reactions
- Some drugs undergo both*
- Some undergo one and not the other
- Phase 1 doesn't necessarily have to come first
- They're not always good - they can produce horribly toxic metabolites
*With regards to paracetamol, these are competitive processes.
That's all we're going to say about metabolism in this post - we'll cover metabolism in all its glory another time.
What actually happens to the drug?
If we inject a single intravenous bolus dose of a drug, and then measure the plasma concentration over time, it generally looks something like this.
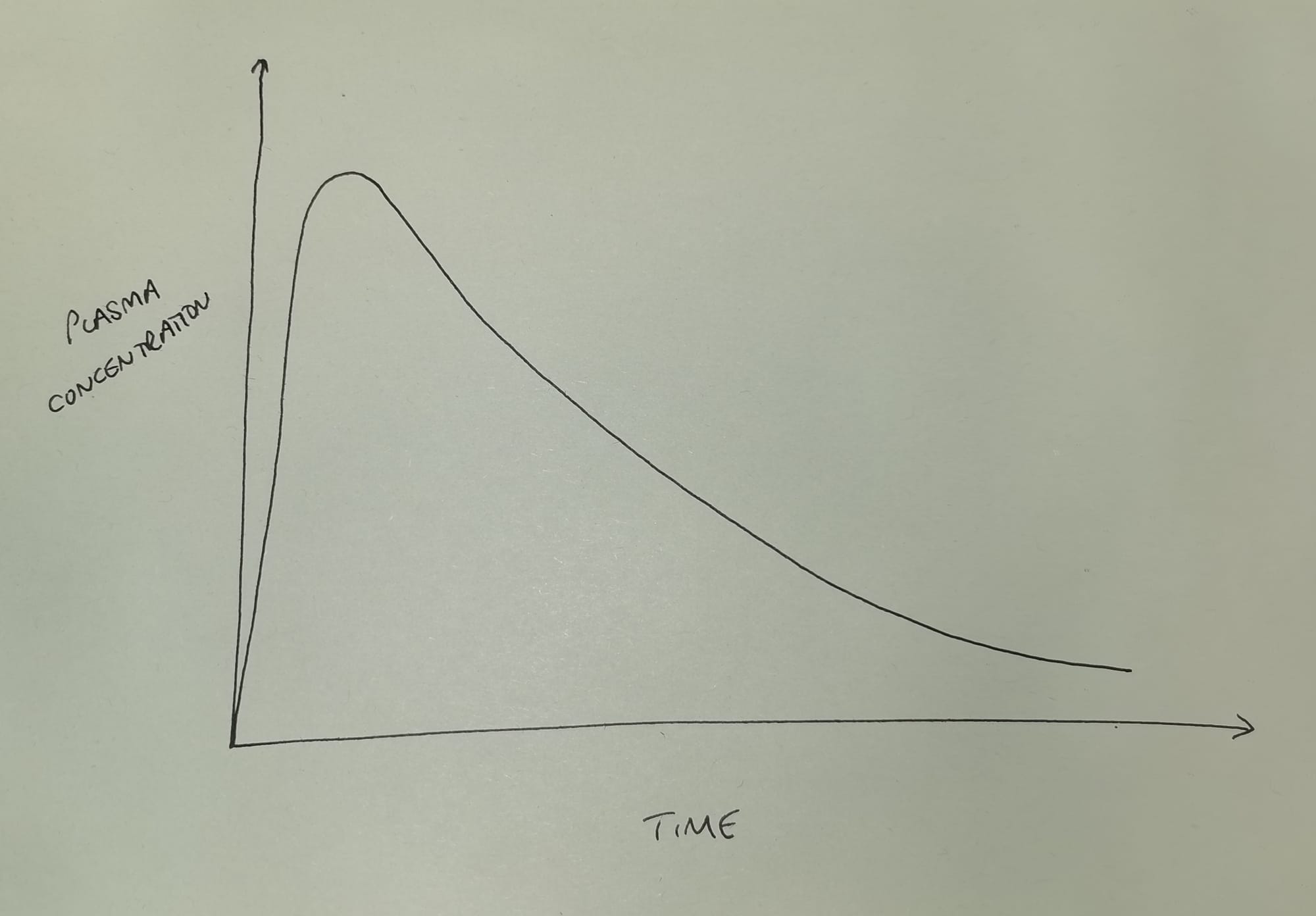
As you can see the plasma concentration initially shoots upwards as we inject the drug, and then rapidly starts to fall in an exponential manner as the drug is eliminated (and redistributed*) from the plasma.
- *for the time being, please ignore redistribution, and assume the body is a single compartment of fluid with the drug evenly distributed throughout - it'll allow us to focus on elimination, and generally make everyone's day so much easier.
Here, 'exponential' means that the gradient is becoming less steep as time goes on, and we have an equation for it.
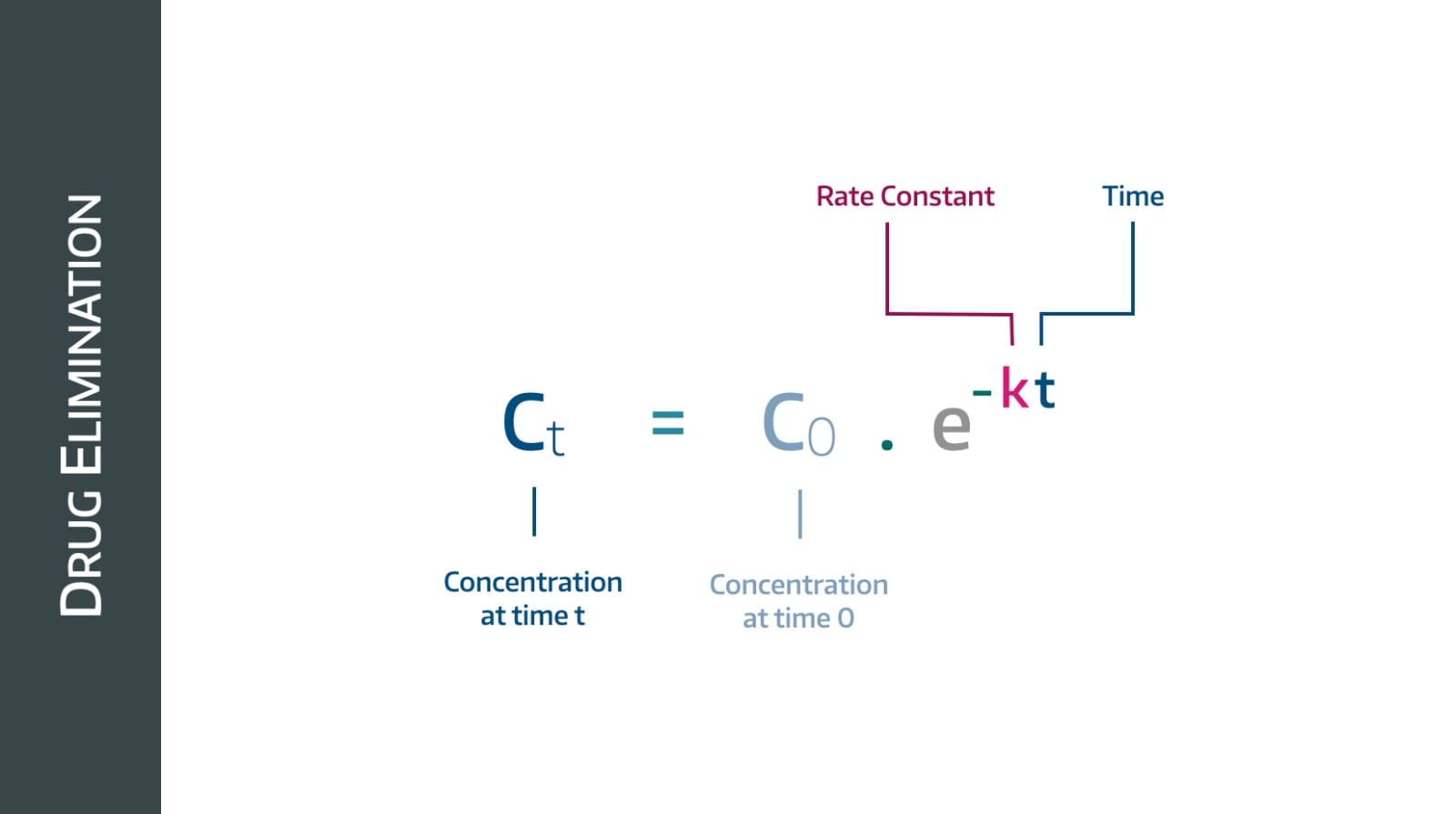
First order kinetics
This behaviour is called first order kinetics, and the vast majority of drugs follow this pattern.
The gradient here - by definition - is (change in plasma concentration)/time and in simple terms tells us the rate of drug removal in mg per minute.
Therefore we can see that the rate at which drug is removed over time is decreasing, which makes sense - to begin with there are loads of drug molecules for the kidneys to remove, and as their concentration drops, there are fewer available, so it takes longer.
What is special about the shape of this graph is this:
- The same proportion of drug is removed per unit time
- i.e. the plasma concentration drops by the same percentage over any given time period
Unfortunately this graph isn't a particularly helpful shape for doing calculations with, so we undertake some mathematical wizardry (we log it) and we get something like this:
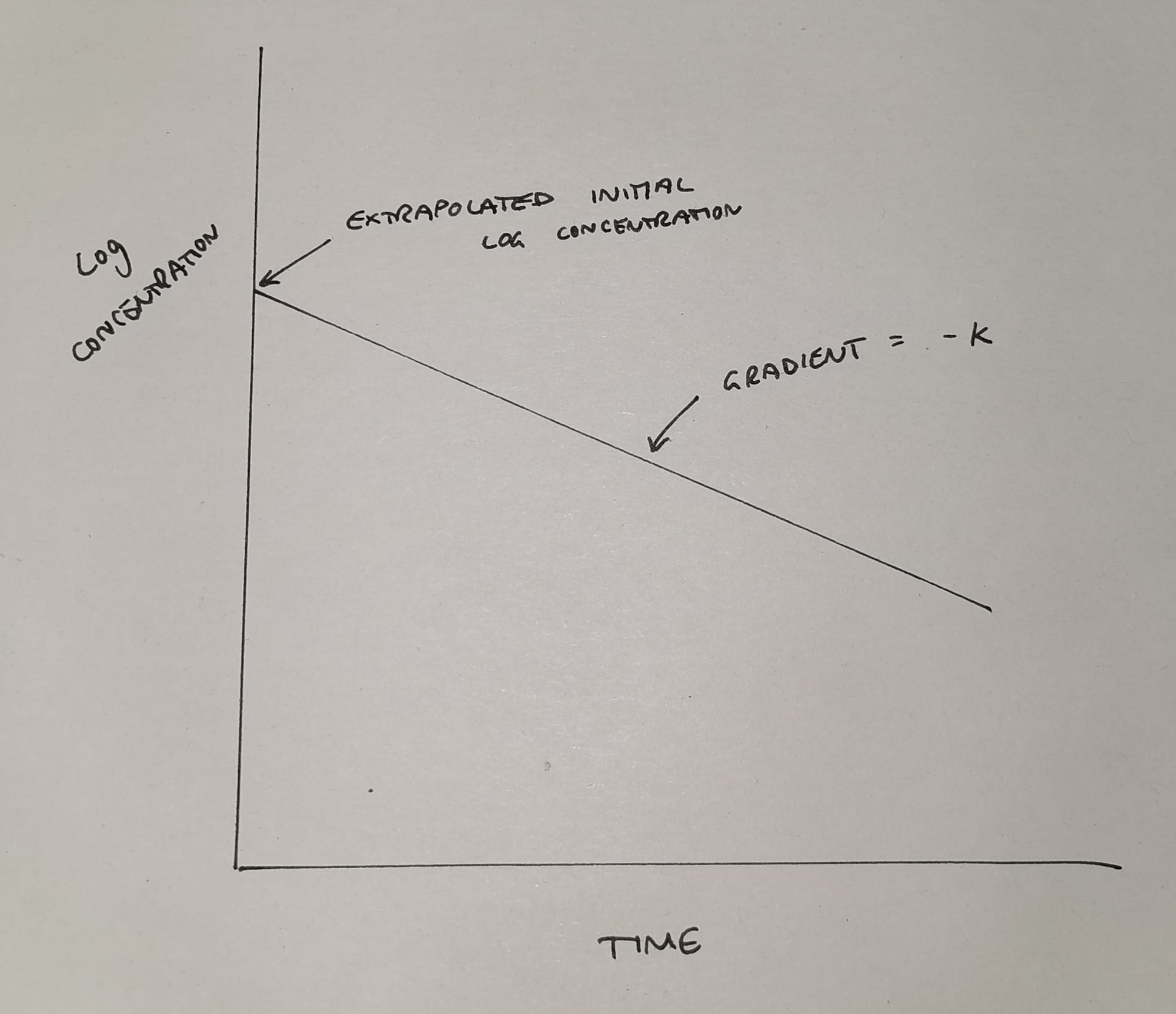
This much simpler graph now tells us two things:
- It allows us to work out the theoretical* initial plasma concentration
- It shows us the elimination rate constant k (the gradient of the slope)
*It's theoretical because it never actually happens - the drug starts being cleared from the blood while we're still injecting - but we like nice neat equations so we make it up using maths.
Now let me introduce you to k - the elimination rate constant.
You only need to know three things
There are three key concepts to understand when discussing elimination of a drug from the plasma:
- The elimination rate constant (k)
- The elimination time constant (τ pronounced 'tau')
- Half life (t1/2)
The elimination rate constant
- k is the proportion of drug eliminated from the plasma over time
- So a k of 0.1 means the plasma concentration drops by 10% per minute
- This gives us that negative exponential graph we saw at the beginning
This makes logical sense, because as the plasma concentration gets smaller, that 10% being removed per minute gets smaller too.
The elimination time constant
- This is simply the reciprocal of the rate constant (1/k)
- This tells us how long it takes for the plasma concentration to drop by 63%
It answers the question "What if the initial rate of elimination didn't slow down, and just kept going at that steepest gradient all the way to the x axis?"
Hence we draw a tangent from the steepest point of the curve, down to the X axis.
We explain this further in the video below - but please don't waste time on this constant, you just need to appreciate that it's 1/k and that t1/2 = 0.693 τ
Half life
- This is the time taken for the plasma concentration to drop by 50%
Look at the plasma concentration graph in the image above and video below, and you'll notice the characteristic shape of exponential decline - meaning that as plasma concentration drops, so too does the rate at which it drops.
This makes sense - assuming your enzymes aren't overwhelmed - because the more molecules there are available to remove, the quicker the rate of elimination is going to be.
As the molecules become fewer and further between, the enzyme has to wait longer for another molecule to come along.
(We're repeating ourselves here, but only because sometimes that's rather helpful...)
If you start at any point on this graph, and measure how long it takes for the plasma concentration to halve from this initial value, it always takes the same amount of time - that's the half life.
- After 1 half life, 50% of the drug is gone
- After 2 half lives, 75% is gone
- After 3 half lives, 87.5%
- After 4 half lives, 93.75%
- After 5 half lives, 96.875%
You get the idea - we generally consider a process 'complete' after five half lives.
- Hopefully it makes sense now that doubling the initial dose of a drug will therefore increase its duration of action by one half life
Why does this matter?
Well it affects how we give our drugs.
All drugs have a therapeutic window - a range of concentrations wherein they have a useful clinical effect. Below this they're useless and above it they're potentially toxic.
If a drug has a very short half life, with a rapid decline in plasma concentration, then it's not going to remain within its therapeutic window for very long. As a result, we'd have to give much more frequent doses to achieve a continuous clinical effect.
These drugs are better off given as continuous infusions.
Especially if they have a narrow therapeutic window with a higher risk of toxicity when given as a bolus dose.
A quick bit about zero order kinetics
We mentioned earlier that most drugs follow first order kinetics.
Compare the above graphs to this one:
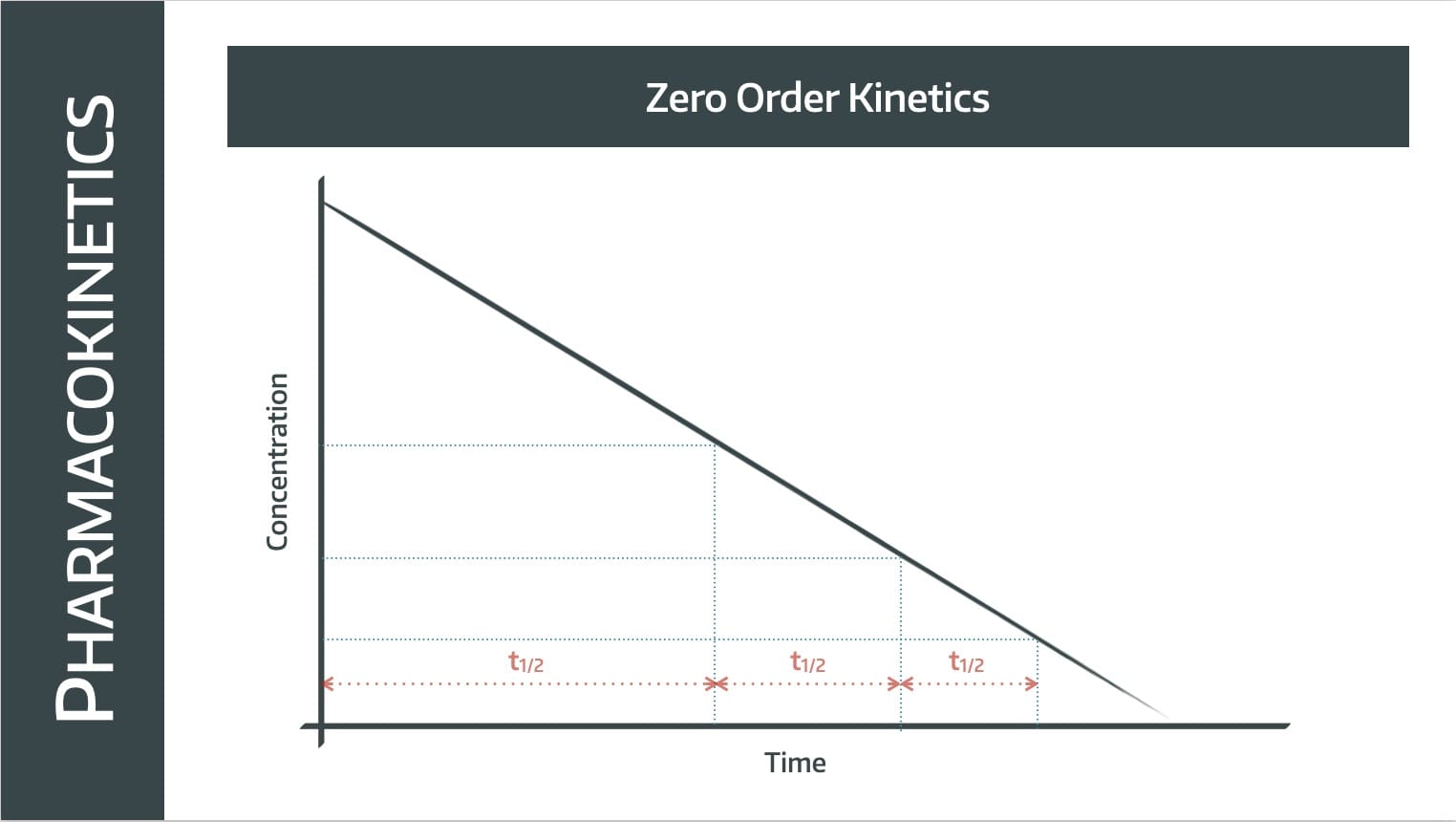
Note this is not a graph that we've log-ed, it's a concentration-over-time graph, which we'd usually expect to be a negative exponential.
In this situation however, the graph is a straight line with a constant gradient, meaning the rate of elimination is constant.
The same number of molecules are removed per unit time, so it takes twice as long to get from 100% to 50% as it does from 50% to 25%.
The half life gets shorter as plasma concentration decreases.
Zero order kinetics take place when the plasma concentration is no longer the limiting factor on rate of elimination, and this generally is seen in two circumstances:
- Massive overdose, with the enzymes completely overwhelmed (a.k.a. saturation kinetics)
- Specific weird drugs that do their own thing
The specific weird drugs in question
- Alcohol - plasma level generally drops by 1 unit per hour
- Phenytoin - easily saturates the enzymes and switches from first to zero order kinetics
- Aspirin
- Thiopentone
The clinical importance of zero order kinetics is that only very small increases in administered dose can lead to dramatic and potentially harmful increases in plasma concentration.
That's quite enough about zero order kinetics I think.
If you've made it this far, then well done.
So far we've said:
- Elimination is the irreversible removal of drug from the plasma
- Most drugs follow first order kinetics
- This means their plasma concentration decreases exponentially over time
- Some drugs are weird and do their own thing
Now let's entertain the mad idea that we might actually want to measure elimination.
Rate of elimination
On the surface, rate of elimination would be a very sensible thing to measure - how much drug in mg or mmol is removed from the plasma per minute?
When we say 'removed' we include both physical removal of the molecule into urine, bile or faeces, or metabolism of the molecule to inactive metabolites.
As we mentioned earlier, we don't include redistribution into fat or other compartments, because those molecules can, and will, flood back into the plasma at some point later on.
Elimination means gone for good.
So when examining this process it initially makes logical sense to think about it in terms of 'amount of drug removed per minute'.
But we run into a problem
The problem here is that this number is going to change depending on the plasma concentration of the drug - as we mentioned earlier.
When there's a very large number of molecules hanging around, they can be eliminated very quickly - assuming your enzymes and transporters haven't reached maximum capacity and been overwhelmed - but as drug plasma concentration drops, so too does the rate at which its molecules are removed.
This means that rate of elimination is going to vary with plasma concentration.
And so that's why we use clearance instead.
Say hello to Clearance
At first glance, this seems like a strange metric to use to measure the efficiency of drug elimination, but the reason we like it is because it remains constant regardless of plasma concentration.
To do this, it takes into account the volume into which the drug is distributed, and it give us the following equation:
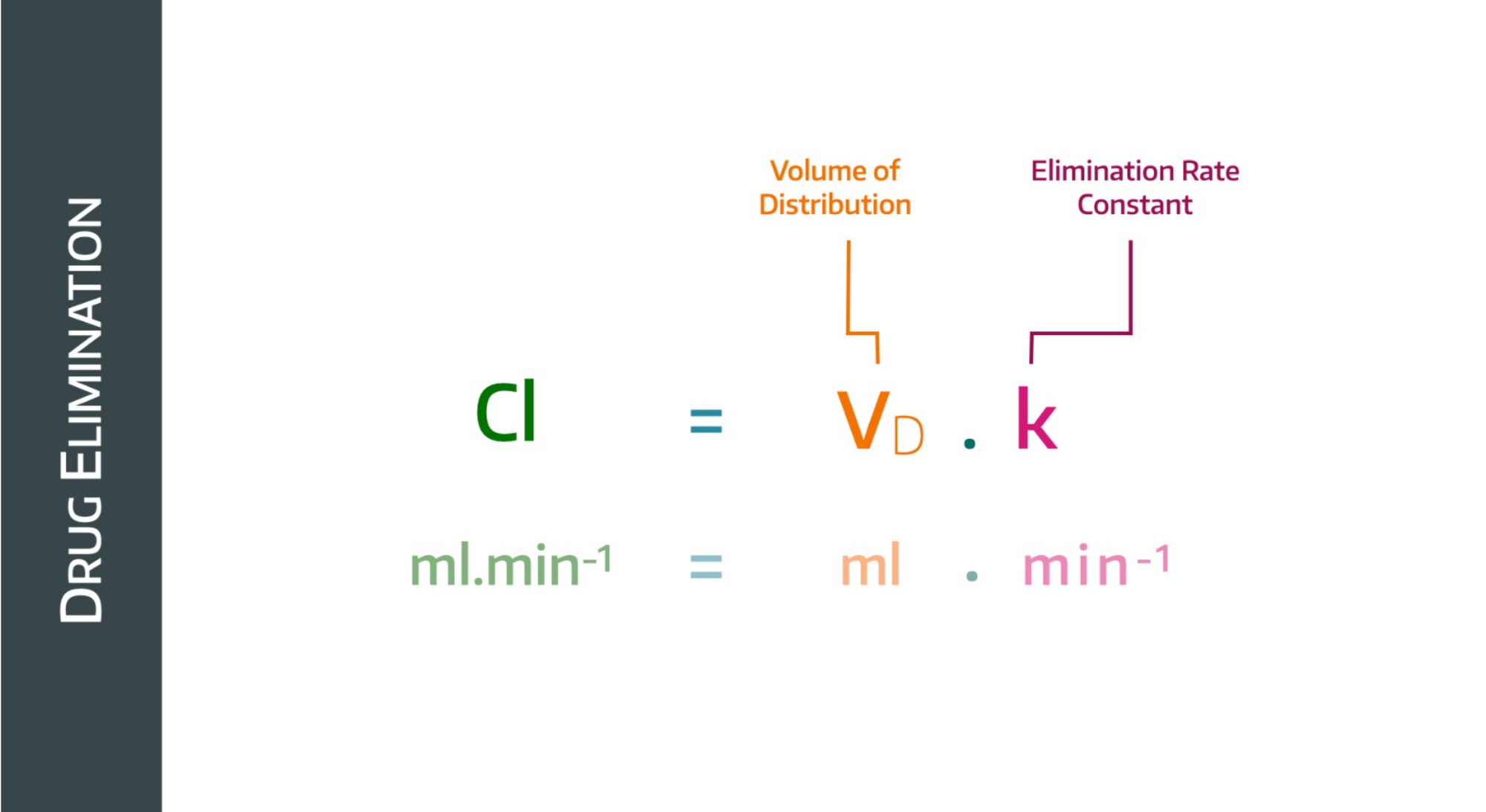
If the plasma concentration drops, then the rate constant will drop too, but the volume of distribution will increase (because a lower concentration implies a higher volume of distribution)
Clearance is therefore essentially the proportionality constant linking rate of elimination to plasma concentration.
Why does this matter clinically?
- We can see here that clearance will be affected by volume of distribution
- Therefore any process or disease that affects volume of distribution, will in turn affect how the drug behaves, particularly its duration of action
- Renal failure can increase volume of distribution via fluid overload
- The endothelial dysfunction of sepsis means previously plasma-bound drugs leak out into the interstitium
- Hypoalbuminaemia leads to reduced protein binding and increased plasma concentration
There are types of clearance
This bit is very simple - the total clearance for the body is a sum of all the separate little bits of clearance carried out by different organs.
- Unsurprisingly, renal clearance tends to be the biggest
- Hepatic clearance is also important
- Other organs are usually forgotten about
This is important to know, because if a drug is predominantly renally cleared, then we need to adjust the dose in renal impairment, whereas if a lot of the work is done by the liver, we probably don't need to worry quite so much.
Alternative ways to calculate clearance:
Why do I care about rate of elimination or clearance?
Because if you want to give an infusion at a steady plasma concentration, you need to know how quickly to infuse the drug.
- Maintenance infusion rate (mg/min) = elimination rate (mg/min)
It's hard to measure the elimination rate, but you can calculate it if you know the clearance and the plasma concentration.
If you give a drug at a steady infusion rate, it will take five half lives to reach steady state at the target plasma concentration (or 96.875% of it at least).
Therefore we usually give a loading dose - which is calculated as follows:
What on earth is context sensitive half life?
Good question.
The graphs we have seen so far generally relate to a single bolus dose of a drug, but things get a little more complex when we start dealing with longer term infusions.
Let's look at propofol
If you infuse propofol for ten minutes and then switch it off, the patient wakes up fairly promptly. If you infuse it for ten hours, the patient takes a lot longer to wake up after you hit stop.
Why?
Propofol accumulates in the fat stores in the body, and then leaches back into the plasma after the infusion stops. This 're-infusion' maintains a certain plasma concentration and prolongs the anaesthetic effect.
The longer the infusion, the longer the effect - the context is simply the duration of the infusion.
Some drugs and their CSHL after 2 hours and 8 hours
- Remifentanil - 5 minutes (2hrs) and 5 minutes (8hrs)
- Propofol - 10 minutes and 40 minutes
- Alfentanil - 30 minutes and 50 minutes
- Thiopental - 100 minutes and 150 minutes
- Fentanyl - 50 minutes and 250 minutes
We call remifentanil context-insensitive - because regardless of the duration of infusion, the half time is always about five minutes.
This is for two reasons:
- It has a smaller volume of distribution, so there's less leeching back in from the fat stores
- It is metabolised by non-specific plasma and tissue esterases, which are so fast and abundant that it's pretty hard to saturate them
Last thing, we promise
Earlier we asked you to bear with us and ignore the whole process of redistribution, and for that we thank you.
Now we can talk about it, and we'll be quick.
The plasma concentration doesn't just drop because of elimination - it also drops because of redistribution, which we've neglected until this point.
This is a graph you need to recognise and understand.
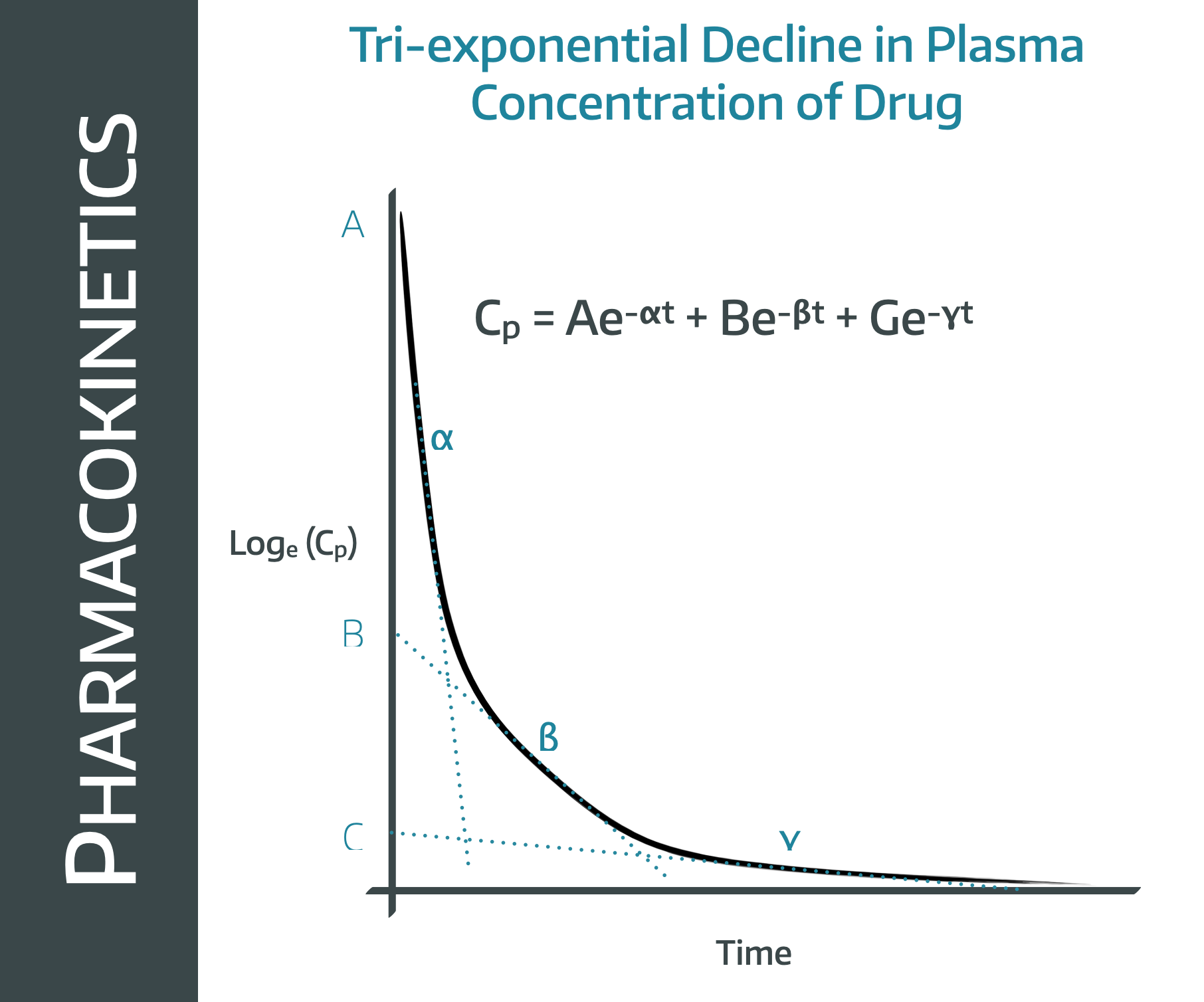
If we look at this graph, it looks a bit like three different curves stuck together, and that's because it is.
Note that this is a log concentration graph, so we'd expect a straight line if it were simple first order kinetics elimination graph.
Instead we have what looks like three straight lines bolted onto one another.
This is because there are three processes going on at the same time, all of which contribute to the fall in plasma concentration over time.
- The drug redistributes into the tissues (the alpha line)
- The drug is eliminated from the plasma (beta)
- The redistributed drug then returns from the tissues into the plasma as we warned it would (gamma)
The first phase gives a very rapid drop in plasma concentration as there is an enormous concentration gradient between plasma and tissues, so lots of molecules redistribute into the tissues fairly quickly.
The second phase represents a steady drop in plasma concentration as the liver and kidneys do their thing. This is the straight line graph from earlier that we've spent pretty much the whole post yapping on about.
In the third phase, the liver and kidneys are still doing their thing, but because the plasma concentration is now lower than the tissue concentration, the molecules start returning to the plasma from the tissue.
This 'autotransfusion' of molecules means the plasma concentration continues to decline, but at a slower rate, hence the gradient isn't quite so steep.
We'll stop talking now.
Useful Tweets and resources
If you want a great video on the reasoning behind the Michaelis-Menten equation, check out this work of art from Andrey K:
Did you know that drug clearance is heavily affected by VV and VA #ECMO: take into account circuit and ECMO factors when evaluating #pharmacokinetics of #antibiotics as explained on this great infographic by 👉 @totalbodydolor 👍 #BEACH2018 #IFAD2018 #FOAMcc #FOAMed pic.twitter.com/TWgPEbHeXI
— Fluid Academy (@Fluid_Academy) November 12, 2018
References and Further Reading

 Alex Yartsev
Alex Yartsev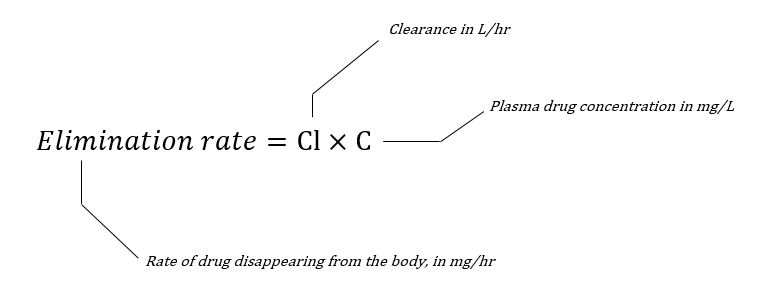
Primary FRCA Toolkit
Members receive 60% discount off the FRCA Primary Toolkit. If you have previously purchased a toolkit at full price, please email anaestheasier@gmail.com for a retrospective discount.

Discount is applied as 6 months free membership - please don't hesitate to email Anaestheasier@gmail.com if you have any questions!
Just a quick reminder that all information posted on Anaestheasier.com is for educational purposes only, and it does not constitute medical or clinical advice.